


También conocido como: factores de la coagulación sanguínea, factores procoagulantes, factor I, factor II, factor V, factor VII, factor IX, factor X, factor XI, factor XIII
Nombre sistemático: actividad de los factores de la coagulación
Aspectos generales
¿Por qué hacer el análisis?
Los factores de la coagulación son proteínas producidas por el hígado y los vasos sanguíneos necesarias para la formación de coágulos en la sangre para evitar sangrados. Este análisis se hace para determinar si la cantidad o actividad de uno o varios de estos factores necesarios es normal, está aumentada o disminuida produciendo alteraciones en la formación de los coágulos.
¿Cuándo hacer el análisis?
Las recomendaciones para solicitar esta prueba son:
- Cuando se ha presentado un sangrado prolongado o inexplicable, y sangrados abundantes en cirugías menores o presencia de hematomas sin traumatismos.
- Cuando los resultados de las pruebas del tiempo de protrombina (TP) y ratio internacional normalizado (INR) o del tiempo de tromboplastina parcial (TTP, aTTP) están alterados sin justificación.
- En los familiares de un paciente con un déficit hereditario de un factor de la coagulación
- Cuando el médico quiere conocer con exactitud la gravedad del déficit de un factor
- Para el seguimiento de la eficacia de un tratamiento
¿Qué muestra se requiere?
La determinación se realiza a partir de una muestra de sangre venosa del brazo.
¿Es necesario algún tipo de preparación previa?
Para esta prueba no se necesita ninguna preparación especial.
¿Qué es lo que se analiza?
La coagulación sanguínea es un proceso complejo en el que participan las plaquetas y numerosos factores de coagulación que tiene como objetivo sellar los vasos sanguíneos dañados para detener el sangrado. Cuando sus concentraciones son bajas, la coagulación sanguínea puede verse comprometida, dando lugar a sangrados inexplicables. La medición de estos factores permite al médico determinar la causa de un sangrado y decidir el tratamiento.
Generalmente, los factores de la coagulación se evalúan midiendo su actividad en la sangre mediante diferentes pruebas de laboratorio. Los ensayos de actividad pueden indicar si la concentración de estas proteínas está disminuida o si no funcionan adecuadamente porque su función está reducida. Con menor frecuencia, puede medirse la concentración de antígeno de los factores de coagulación, que es directamente la cantidad del propio factor. Las pruebas de antígenos de los factores de la coagulación permiten conocer en qué cantidad están presentes esas proteínas, pero no es posible saber si su función es normal. Son pruebas fundamentales para el diagnóstico de deficiencias.
Cuando se produce un sangrado, por ejemplo, después de hacerse una herida, se activa el sistema de la coagulación, taponando el orificio por donde se producía el sangrado mediante un coágulo. El sistema de la coagulación consiste en una serie de proteínas llamadas factores de la coagulación, que se activan de manera secuencial por un mecanismo conocido como cascada de la coagulación. El resultado final consiste en la formación de unas hebras de fibrina insolubles que se entrecruzan en el lugar de la lesión, junto con unos fragmentos celulares conocidos como plaquetas, formándose así un coágulo estable. Este coágulo impide las pérdidas adicionales de sangre y permanece allí hasta que el área ha cicatrizado.
Por otra parte, hay que tener en cuenta que la coagulación es un proceso dinámico. Una vez formado el coágulo, se activan otros factores para limitar el aumento de tamaño del coágulo o disolverlo mediante un mecanismo conocido como fibrinólisis. El coágulo se elimina con el tiempo, a medida que la lesión se va curando. En las personas sanas, este equilibrio entre formación y eliminación del coágulo asegura que no se produzcan sangrados excesivos, y que los coágulos se vayan eliminando cuando ya no son necesarios.
En los pacientes con trastornos hemorrágicos, el proceso de la coagulación no funciona correctamente porque tienen poca cantidad de plaquetas o de factores de coagulación, o bien porque no funcionan correctamente. Existe una gran variedad de trastornos hemorrágicos, algunos de los cuales son hereditarios (se transmiten de una generación a la siguiente de la misma familia), mientras que otros son adquiridos después del nacimiento. Si una persona tiene signos o síntomas de alguna de estas enfermedades, se puede realizar un análisis de los factores de coagulación, para establecer el diagnóstico y el tratamiento de elección.
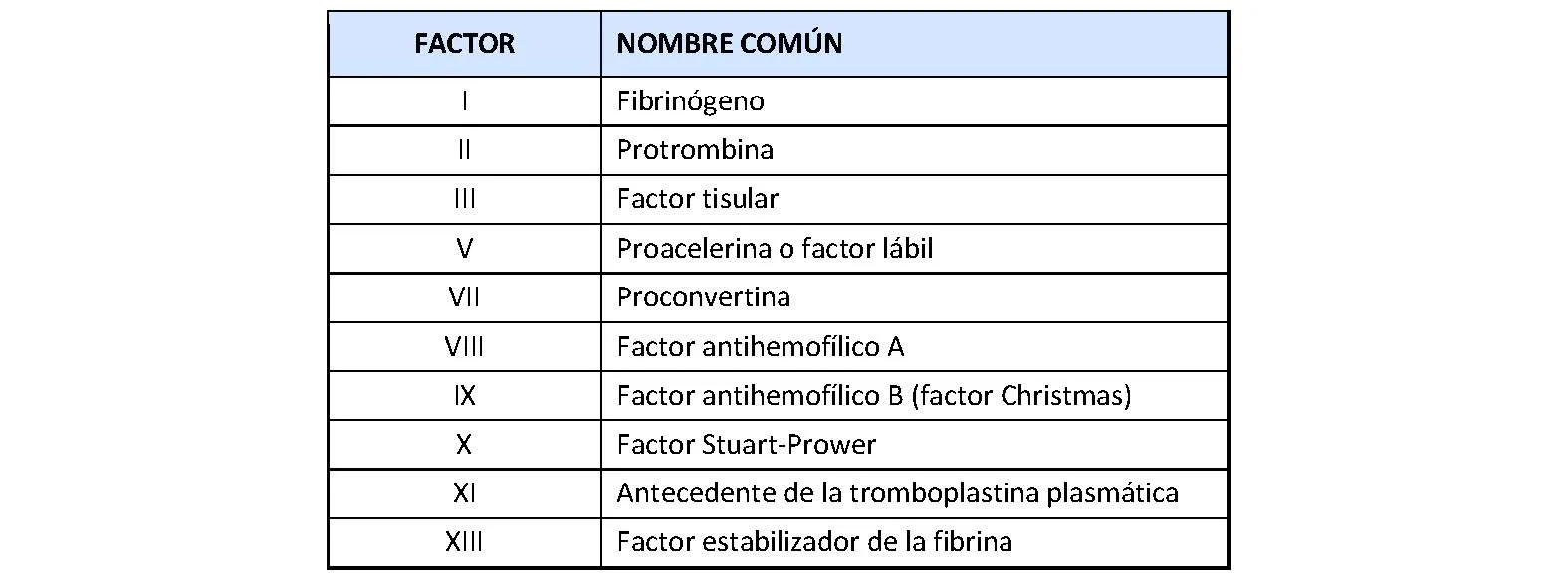
Existen nueve factores proteicos de la coagulación que se analizan de manera rutinaria A estos factores se les asigna un número romano y, en ocasiones, también un nombre. Cada uno de ellos tienen una función diferente en la cascada de coagulación y todos son imprescindibles para la formación del coágulo.
- Factor I o Fibrinógeno: Precursor soluble de la fibrina, que es la base estructural del coágulo.
- Factor II o Protrombina: Se transforma en trombina, es una de las enzimas centrales de la coagulación.
- Factor III o Factor tisular: Necesario para iniciar la vía extrínseca de la coagulación.
- Factor V o Proacelerina: Es un cofactor del factor Xa.
- Factor VII o Proconvertina: Participa de manera conjunta con el Factor tisular en la activación de la vía extrínseca.
- Factor VIII o Factor antihemofílico A: También es un cofactor del Xa.
- Factor IX o Factor antihemofílico B (factor Christmas): De manera conjunta con el factor VIII participa en la activación del factor X.
- Factor X o Factor de Stuart-Prower: Es el factor que inicia la vía común y permite la transformación de la protrombina a trombina.
- Factor XI o Antecedente de la tromboplastina plasmática: Participa en la amplificación de la vía intrínseca.
- Factor XIII o Factor estabilizador de la fibrina: Permite la estabilización de la fibrina para la formación del coágulo.
Preguntas comunes
¿Cómo se utiliza?
El análisis de los factores de la coagulación se realiza para determinar si su actividad es suficiente para controlar el proceso de la formación del coágulo. Sirve para determinar si la concentración de uno o varios factores está disminuida, o los factores están ausentes, es decir, por debajo de los límites de detección del método de medida, y por lo tanto existe una formación inadecuada del coágulo que puede dar lugar a sangrados. Estas pruebas también permiten saber si estos factores están aumentados y por ello puede existir una formación excesiva de coágulos (trombosis) que pueda llegar a bloquear la circulación de la sangre (tromboembolismo).
Para evaluar la función de un factor concreto, se puede medir su actividad. Si se encuentra una disminución de la actividad puede medirse el antígeno, para establecer si la baja actividad hallada es debida a una cantidad escasa del factor, o bien a un defecto de la función del factor evaluado.
A veces se miden los factores de la coagulación en las personas con antecedentes familiares de sangrados.
¿Cuándo se solicita?
Las pruebas de los factores de la coagulación suelen solicitarse cuando el paciente presenta un tiempo de protrombina (TP) y ratio internacional normalizado (INR) o un tiempo de tromboplastina parcial (TTP, aTTP) prolongados sin justificación. Estas pruebas se solicitan como pruebas de cribado, para determinar si existe algún problema relacionado con la coagulación en los pacientes con signos o síntomas de un trastorno hemorrágico, como pueden ser la formación de hematomas, sangrado de las encías, sangrado excesivo por cortes pequeños o epistaxis (sangrado nasal) frecuentes.
Los factores de la coagulación también pueden solicitarse cuando se sospecha que existe una condición adquirida que está originando los sangrados, como una enfermedad hepática que produciría una síntesis disminuida de los factores de coagulación o un déficit de vitamina K.
El estudio de los factores de la coagulación puede realizarse cuando se sospecha una deficiencia hereditaria de un factor, como la enfermedad de von Willebrand o la hemofilia A, especialmente si los episodios de sangrado se producen a edades tempranas o cuando se sabe que un familiar presenta una deficiencia de un factor. Si se sospecha un déficit hereditario, puede realizarse el estudio al resto de los familiares, para confirmar el diagnóstico y establecer si existen familiares portadores del déficit, o incluso si hay familiares que tengan el mismo déficit, pero de manera más leve o totalmente asintomática.
A veces se realiza el estudio a las personas que tienen una deficiencia conocida de algún factor, para el seguimiento y evaluación de la eficacia del tratamiento.
En ocasiones, también se puede realizar la prueba en los pacientes con un exceso inexplicable en la formación de coágulos (trombosis), para determinar si existe una concentración anormalmente elevada de algún factor (por ejemplo: el aumento de la actividad del factor VII está relacionado con una coagulación excesiva).
¿Qué significa el resultado?
Una actividad normal de los factores de la coagulación suele indicar que la coagulación es normal. Una actividad baja de uno o varios factores de la coagulación suele indicar que existe algún problema con la capacidad de coagulación. Cada uno de los factores de la coagulación debe estar presente en cantidades suficientes para permitir un proceso normal de la coagulación, aunque la cantidad necesaria varía de un factor a otro. Los resultados suelen expresarse en forma de porcentaje, considerándose normales los valores del 100%. Por ejemplo, un factor VIII del 30% es un resultado anormalmente bajo.
Los déficits de los factores de la coagulación pueden ser hereditarios o adquiridos (secundarios a otras causas), moderados o graves, y permanentes o transitorios.
Si están afectados dos o más factores, es probable que se trate de una alteración adquirida. Las deficiencias adquiridas son raras y pueden estar causadas por patologías crónicas o agudas, entre ellas:
- Exceso de coagulación: produce el consumo de los factores, por ejemplo, la coagulación intravascular diseminada.
- Enfermedad hepática: por ejemplo, la cirrosis.
- Algunos tipos de cáncer.
- Exposición a ciertos tipos de veneno de serpiente.
- Malabsorción de grasa.
- Déficit de vitamina K.
- Tratamiento anticoagulante.
- Transfusiones masivas: por ejemplo, cuando se transfunden exclusivamente hematíes.
Los trastornos hereditarios son raros y tienden a implicar solamente un factor, que puede estar ausente (inferior al límite de detección del método de medida), o disminuido.
Las hemofilias A y B son el ejemplo característico de un trastorno hereditario. Se trata de deficiencias de los factores VIII y IX ligadas al cromosoma X, que tienen lugar casi exclusivamente en varones. Las mujeres suelen ser portadoras asintomáticas o presentar sangrados leves. Existen otras deficiencias hereditarias de factores de la coagulación, no asociadas al cromosoma X, que se dan por igual en hombres y en mujeres.
La gravedad de los síntomas asociados al déficit hereditario de un factor depende del factor afectado, de la cantidad que queda disponible y de si está o no funcionando correctamente. Los síntomas pueden variar de episodio a episodio, desde un sangrado después de la manipulación dental, hasta sangrados recurrentes en zonas como las articulaciones o los músculos.
Es posible que las personas con deficiencias moderadas presenten muy pocos síntomas y sean diagnosticados ya cuando son adultos, después de un traumatismo o una intervención quirúrgica, o si se observan resultados anormales del tiempo de protrombina (TP) y ratio internacional normalizado (INR) o del tiempo de tromboplastina parcial (TTP, aTTP).
Las personas con deficiencias graves de un factor pueden presentar su primer episodio de sangrado muy precozmente; por ejemplo, un bebé varón con un déficit de factor VIII, IX o XIII puede sangrar excesivamente después de la circuncisión.
Un aumento de varios factores simultáneamente puede surgir en distintas situaciones agudas, estrés o inflamación. Por ejemplo, el fibrinógeno es un reactante de fase aguda. Sin embargo algunas personas presentan de manera persistente aumentos de ciertos factores, como el factor VIII, que pueden asociarse a un mayor riesgo de trombosis venosa.
¿Hay algo más que debería saber?
Una vez identificado el factor deficitario, este puede administrarse de forma artificial. Esto puede conseguirse mediante la transfusión de plasma normal, que contenga todos los factores que faltan, o bien mediante un concentrado, o un factor adicional (algunos se comercializan en forma recombinante, como el factor VIII). Estos tratamientos pueden utilizarse durante los episodios de sangrado, o de manera preventiva para conferir protección frente a un sangrado excesivo durante una próxima intervención quirúrgica o una manipulación dental.
¿Qué es el factor von Willebrand?
El factor von Willebrand es el responsable de permitir que las plaquetas se adhieran a la pared vascular lesionada. También es la proteína de transporte del factor VIII. Un déficit de factor von Willebrand puede originar la enfermedad de von Willebrand, un trastorno hemorrágico hereditario. Aunque el factor von Willebrand suele solicitarse juntamente a otros factores de la coagulación si se sospecha un déficit hereditario, normalmente se le considera separadamente, debido a que está más asociado a las plaquetas y no forma parte directa de la cascada de la coagulación.
¿Qué es el factor V de Leiden?
El factor V de la coagulación, conocido como proacelerina, es una proteína que participa en la cascada de coagulación en la conversión de protrombina a trombina por el factor Xa en la superficie de las plaquetas. Su unión a este factor Xa aumenta la velocidad de generación de trombina, que es fundamental para la formación eficiente y rápida del coágulo. Su funcionamiento está regulado por le proteína C activada, esta es una proteína antitrombótica que lo que hace es desactivar a este factor V y detener la formación del coágulo.
El factor V de Leiden es una mutación genética hereditaria (que se transmite en las familias) que altera a este factor V de la coagulación. Esta mutación lo que provoca es que sea menos sensible a la proteína C, es decir, que no puede llegar a desactivarlo. Esto hace que aumente el riesgo de formación de coágulos (trombos) y que se desarrollen episodios de trombosis venosa profunda (TVP) o embolismo pulmonar (EP). Es un factor de riesgo que debe considerarse en situaciones como embarazo, uso de anticonceptivos orales, cirugía o inmovilidad prolongada, ya que también son factores de riesgo de la formación de coágulos.
¿Por qué algunos trastornos hereditarios de la coagulación son más graves que otros?
La gravedad del sangrado depende del individuo, del grado de disminución de la concentración o de la función del factor anómalo, así como del tipo de factor deficitario. Los individuos con un déficit grave de un factor, o bien con un factor con muy poca actividad, presentan manifestaciones más graves de la enfermedad. Las personas que tienen una copia normal del gen y otra copia alterada (heterocigotos) tienden a presentar sangrados menos graves que las que tienen las dos copias alteradas (homocigotos).
Es posible que una persona con un déficit de factor XII esté asintomática. Este déficit es raro y origina resultados anormales del tiempo de tromboplastina parcial (TTP, aTTP) pero no se asocia a un mayor riesgo de sangrado.
Enlaces
Pruebas relacionadas:
Tiempo de protrombina (TP) y ratio internacional normalizada (INR)
Tiempo de tromboplastina parcial (TTP, aTTP)
Estados fisiológicos y enfermedades:
Estados de hipercoagulabilidad sanguínea
En otras webs:
Federación Española de Hemofilia (Fedhemo): Hemofilia
Manual MSD: Introducción a los trastornos de la coagulación
También conocido como: HSV-1, HSV-2, HHV1, HHV2, VHS, herpes oral, herpes labial, herpes genital
Nombre sistemático: virus del herpes simple tipo 1 y tipo 2
Aspectos generales
¿Por qué hacer el análisis?
Para hacer un cribado o un diagnóstico de la infección por el virus del herpes simple (VHS).
Pregúntenos



Síguenos



Fecha de creación de la nueva página: 1 de enero de 2023
ISSN 2939-8759