


También conocido como: mucoviscidosis
¿En qué consiste?
La fibrosis quística (FQ) es una enfermedad hereditaria que afecta principalmente a los pulmones, páncreas y glándulas sudoríparas. Las personas afectadas producen mucosidades densas y pegajosas que favorecen las infecciones respiratorias y bloquean la liberación de los enzimas pancreáticos, impidiendo la normal digestión de proteínas y lípidos.
La FQ está causada por una mutación en las dos copias (una de la madre y otra del padre) del gen CFTR localizado en el cromosoma 7. Hasta la fecha se han identificado más de 2.000 mutaciones distintas en el gen CFTR, pero solamente unas pocas son las causantes de la mayoría de los casos. La mayoría de los casos de FQ están producidos por una mutación conocida como deleción F508 (DF508).
El gen CFTR es responsable de la correcta producción de la proteína CFTR (regulador de la conductancia transmembrana asociado a la fibrosis quística). En la FQ, esta proteína puede estar ausente o bien no funcionar de manera adecuada. Por ello, el cloruro no puede salir de las células y formar parte de las secreciones y fluidos corporales, por lo que esos fluidos se vuelven densos y pegajosos. Las células más ricas en CFTR son las que recubren el interior de los pulmones, el páncreas, las glándulas sudoríparas y salivales, el intestino y los órganos reproductivos, por lo que son las estructuras más afectadas.
Aunque los signos y síntomas de la FQ varían entre personas, la enfermedad pulmonar y la enfermedad pancreática suelen aparecer en la primera infancia. Esta enfermedad también causa infertilidad por problemas en el desarrollo de los conductos deferentes, que transportan los espermatozoides desde los testículos al exterior.
La FQ está causada por una mutación en las dos copias (una de la madre y otra del padre) del gen CFTR localizado en el cromosoma 7. Ambos alelos de este gen deben presentar la mutación para producir la enfermedad de la FQ. Si únicamente hay un alelo mutado, el individuo será portador de FQ. Los portadores normalmente no presentan ningún signo o síntoma de FQ, pero pueden transmitir su copia mutada del gen a sus hijos.
Acerca de la fibrosis quística
Signos y síntomas
Entre los signos y síntomas más frecuentemente asociados a FQ se encuentran:
- Tos crónica con producción de esputo.
- Infecciones respiratorias de repetición o persistentes, como bronquitis o neumonías.
- Congestión nasal y sinusitis de repetición, pólipos nasales.
- Dolor o malestar abdominal.
- Íleo meconial en recién nacidos.
- Diarrea crónica, de olor nauseabundo y con heces grasientas.
- Pérdida de peso o malnutrición.
- Retraso en el desarrollo y dificultad para ganar peso.
- Disminución de la concentración de proteínas en sangre y aparición de edemas.
La FQ interfiere en el equilibrio hidroelectrolítico del organismo. El sudor de las personas con FQ contiene hasta cinco veces más sal (cloruro sódico) de lo normal. La pérdida excesiva de sodio y cloruro puede afectar al ritmo cardíaco y provocar un estado de shock.
En la FQ, la mucosidad que lubrica los pulmones se vuelve espesa y pegajosa, siendo un caldo de cultivo perfecto para los microorganismos y provocando infecciones respiratorias de repetición. Estas infecciones deben tratarse de manera agresiva con antibióticos intravenosos, orales o inhalados. La mayoría de los problemas asociados a la FQ se deben a infecciones respiratorias y enfermedades pulmonares.
La formación de tapones de moco en el páncreas bloquea los conductos que comunican con el intestino impidiendo el transporte de enzimas pancreáticos para digerir las proteínas y lípidos de la dieta, que por tanto no se pueden absorber (malabsorción). Esto puede provocar deficiencias vitamínicas y malnutrición. La administración oral de suplementos de enzimas pancreáticos y la reposición de las vitaminas liposolubles (A, D, E y K) junto con una dieta pobre en lípidos y rica en proteínas ayudan a mitigar estos síntomas. Si la enfermedad pancreática es suficientemente importante, algunos individuos con FQ pueden desarrollar una diabetes.
Otros problemas asociados a FQ incluyen:
- Formación de piedras en la vesícula biliar (cálculos biliares).
- Pancreatitis.
- Retraso en el crecimiento y retraso de la pubertad.
- Ensanchamiento o forma redondeada de las puntas de los dedos de manos y pies (“dedos en palillos de tambor”).
- Enfermedad hepática crónica y cirrosis biliar.
- Prolapso rectal (protrusión del recto a través del ano).
- Infertilidad.
Pruebas relacionadas
Las pruebas de laboratorio permiten hacer un cribado y diagnosticar la FQ, pero también detectar a los portadores de FQ o valorar a los pacientes ya diagnosticados para ajustar el tratamiento.
Pruebas de cribado y diagnóstico
Aunque es posible realizar una prueba para conocer el estado de portador antes del embarazo, no se suele utilizar excepto en los familiares de pacientes ya diagnosticados de FQ o en las parejas de personas portadoras conocidas.
La FQ es una de las siete enfermedades que se criban en todos los recién nacidos del territorio español, ya que su cribado forma parte de la cartera común básica de servicios asistenciales del Sistema Nacional de Salud.
Se pueden utilizar diversas pruebas para el cribado de FQ, aunque un resultado positivo siempre hay que confirmarlo mediante la prueba de cloruro en sudor.
- Mutación del gen de la fibrosis quística: permite detectar tanto a los pacientes con la enfermedad como a los portadores asintomáticos. Se recomienda el uso de perfiles que puedan detectar las mutaciones más comunes del gen CFTR en función de la procedencia del paciente, aunque las mutaciones raras puede que no se detecten. Si se identifican dos mutaciones causantes de FQ, una en cada uno de los progenitores, conviene que un asesor genético le explique el riesgo relativo que la pareja tiene de concebir un hijo con FQ. Por otra parte, es muy importante ofrecer asesoramiento genético a los familiares de pacientes diagnosticados de FQ y a cualquier persona que se vaya a someter a esta prueba.
- Tripsinógeno: el tripsinógeno se produce en el páncreas y se transporta al intestino donde se activa para formar un enzima denominado tripsina, que participa en la digestión de las proteínas. En la FQ, los tapones de moco pueden obstruir los conductos pancreáticos e impedir el acceso del tripsinógeno al intestino, con lo que se acumula en la sangre.
- Prueba del cloruro en sudor: esta prueba consiste en medir el cloruro en una muestra de sudor recogida de forma especial. Debido a que la proteína CFTR está alterada o ausente y el transporte de cloruro se ve limitado, el sudor en una persona con FQ puede ser hasta cinco veces más salado que el de una persona sin la enfermedad. Esta prueba se considera el patrón de oro (gold standard) para el diagnóstico de FQ, y se suele utilizar para confirmar los resultados positivos obtenidos en otras pruebas.
- Gradiente de potencial nasal transepitelial: esta prueba se usa con menos frecuencia que las anteriores, ya que es difícil de realizar y está disponible en pocos centros. Consiste en la colocación de un electrodo en la nariz que se empapa de diferentes líquidos con sales. Los líquidos modifican el transporte activo de iones como sodio y cloruro a través de la capa de células que recubre el interior de la nariz, generando una diferencia de potencial eléctrico que puede medirse. El transporte anormal de sodio y cloruro en el epitelio respiratorio de las personas con FQ se asocia a un patrón alterado de diferencia de potencial nasal, en comparación con una persona sin FQ.
En la siguiente infografía se resumen algunos datos sobre la fibrosis quística:
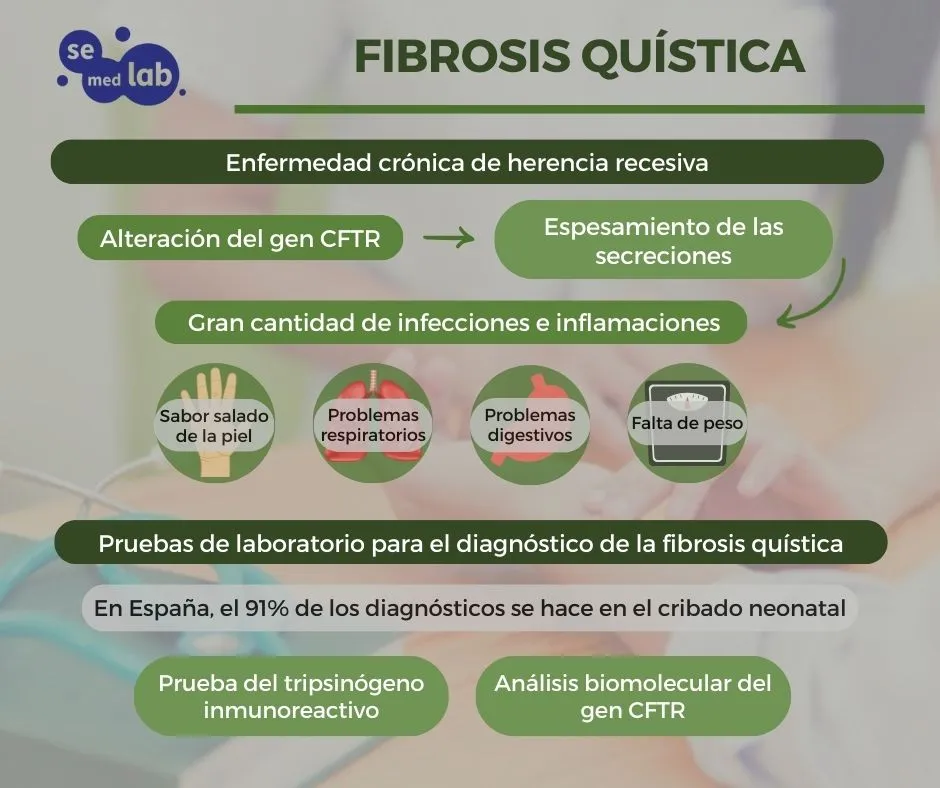
Entre otras pruebas que se utilizan para controlar la función de los órganos, la fertilidad y para detectar infección pulmonar se incluyen:
- Hemograma, bioquímica general y perfil hepático: para evaluar el estado de salud general y la función de los principales órganos, además de detectar infecciones.
- Glucosa y hemoglobina glicada (HbA1c): para detectar los casos de diabetes.
- Amilasa y lipasa: para detectar enfermedades pancreáticas.
- Grasas en heces: para detectar malabsorción o insuficiencia pancreática.
- Estudio del semen: para detectar infertilidad.
- Cultivo de esputo: para detectar infecciones pulmonares.
Otras pruebas diagnósticas ajenas al laboratorio
También se realizan pruebas, que no son de laboratorio, como radiografías de huesos y de tórax, series del tracto gastrointestinal alto e intestino delgado y pruebas de función pulmonar.
Para más información consultar la página RadiologyInfo.org.
Tratamiento
Actualmente no existe cura para la FQ, sino tratamientos para reducir los signos y síntomas y mejorar la calidad de vida. Los tratamientos se establecen a medida para cada paciente y su principal objetivo es favorecer la eliminación del exceso de moco, para prevenir las infecciones pulmonares y las obstrucciones en el tracto gastrointestinal. También es necesario asegurar una nutrición adecuada con dietas especializadas.
Algunas terapias incluyen ejercicios físicos y fisioterapia, además de fármacos como antibióticos, antiinflamatorios y broncodilatadores.
Recientemente se han desarrollado una serie de fármacos denominados moduladores CFTR, cuyo objetivo sería mejorar el funcionamiento de la proteína defectuosa en pacientes con mutaciones concretas.
En ciertas ocasiones se puede realizar trasplante pulmonar en pacientes con FQ, aunque se tienen que calibrar los riesgos de la cirugía y de la inmunosupresión y los posibles beneficios en la duración y calidad de vida de los pacientes.
Se sigue investigando para conseguir un tratamiento que permita curar la enfermedad; en los últimos diez años se ha avanzado considerablemente en las opciones de tratamiento que han proporcionado mejor calidad de vida a los afectados.
Enlaces
Pruebas relacionadas:
Mutación del gen de la fibrosis quística
Estados fisiológicos y enfermedades:
Cribados:
Cribado de los recién nacidos
Asociaciones de pacientes:
Federación Española de Fibrosis Quística-FEFQ
Asociación Andaluza de Fibrosis Quística-AAFQ
Associació Catalana de Fibrosi Quística -ACFQ
Asociación Madrileña de Fibrosis Quística-AMCFQ
Arnasa Asociación de Fibrosis Quística de Euskadi-ARNASA FQ Euskadi
Asociación Navarra contra la Fibrosis Quística-FQNavarra
Asociación Murciana de Fibrosis Quística-AMFQ
Asociación Castellano-Leonesa contra la Fibrosis Quística - FQCYL
En otras webs:
Ministerio de Sanidad: Programa de cribado neonatal
Sociedad Española de Fibrosis Quística (SEFQ)
Federación Española Fibrosis Quística (FEFQ)
KidsHealth: Fibrosis quística
Cystic Fibrosis Foundation: ¿Qué es la fibrosis quística (FQ)?
Sociedad Española de Neumología y Cirugía Torácica (SEPAR): Fibrosis quística
Pregúntenos



Síguenos



Fecha de creación de la nueva página: 1 de enero de 2023
ISSN 2939-8759